Analyzing a Transcription factor from JASPAR, UniProbe, EpiSELEX-seq, or MeDReaders
1.1. Make a Selection: Click on "Search" in the navigation bar on the left and then choose a database to search in.
1.2. Enter Search Criteria: Filter search results with the dropdown menus and/or search by transcription factor name. Searching by name is not case sensitive. Search criteria are optional. Click "Search" to get search results.
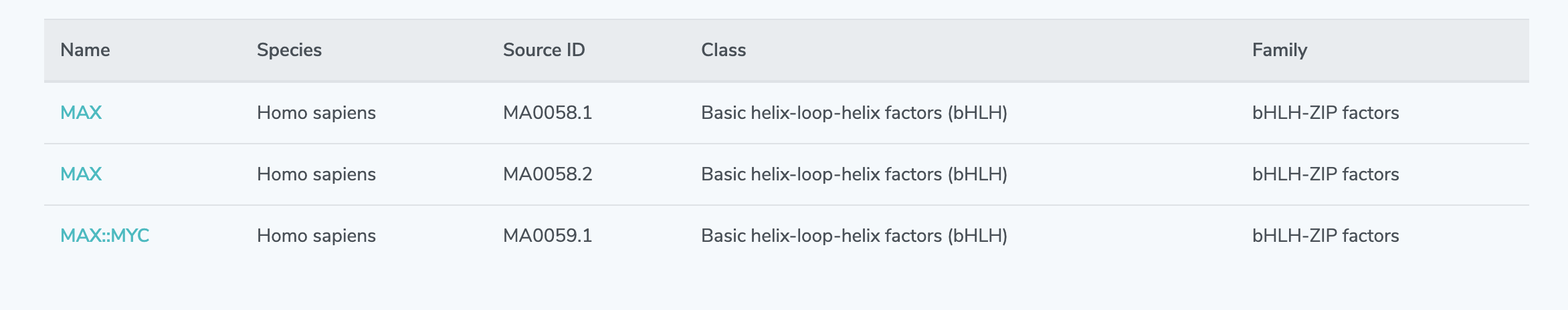
2. Search Results: Browse search results and click on the name of your desired transcription factor. If no results are shown, please broaden your query and search again.
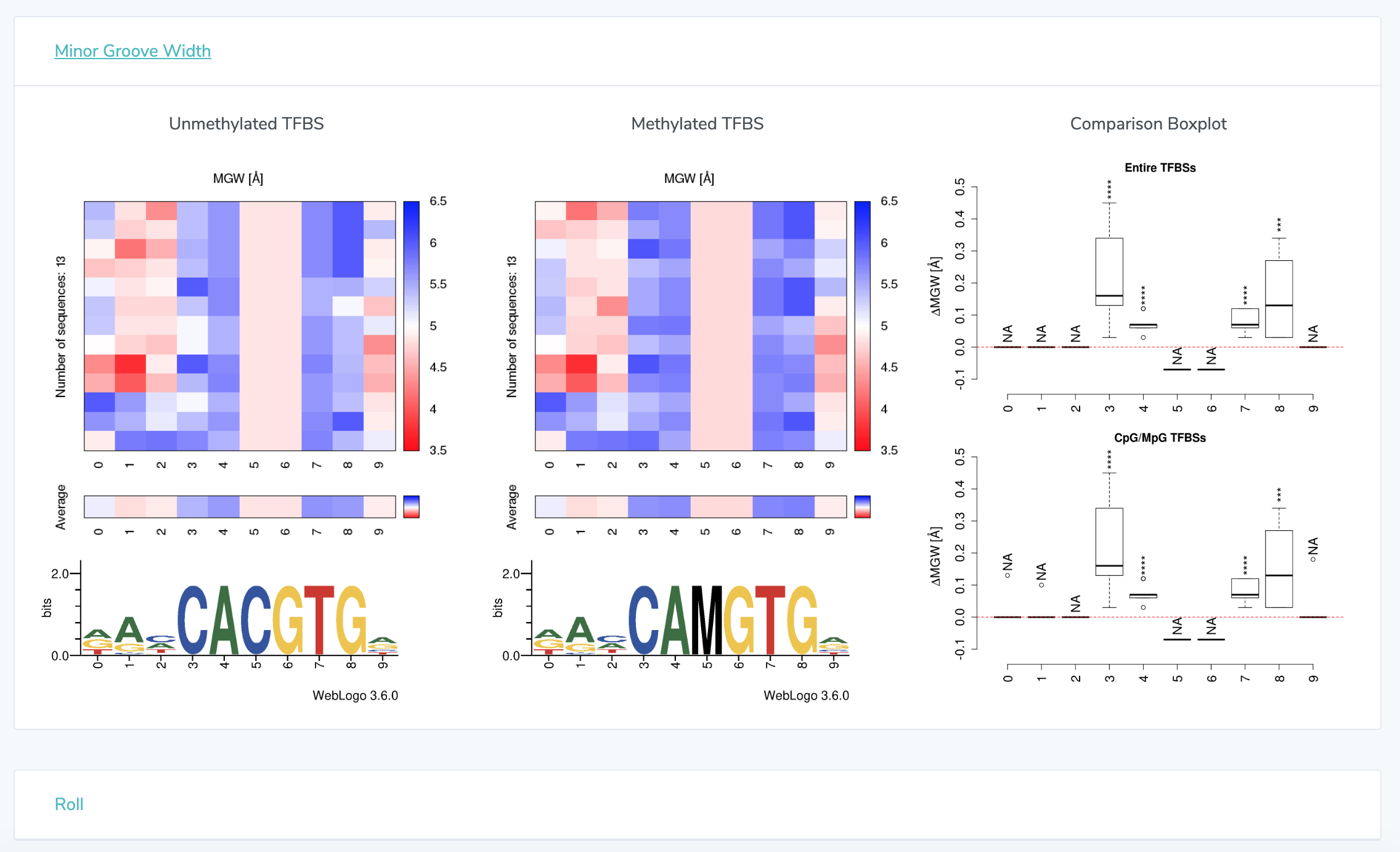
3. Shape Analysis: At the top, detailed information is presented on each transcription factor. This information varies depending on the source. See "UniProbe Detail Page" below for information how to make selections with UniProbe. Click on any of the 13 feature names to view heatmaps representing shape features. Averages for each base position are shown below the heatmaps. For Minor Groove Width, Roll, Propeller Twist, and Helical Twist, heatmaps for the methylated sequence are shown adjacent to unmethylated heatmaps. Sequence logos are displayed below heatmaps.
UniProbe Detail Page
1. PWM Selection If multiple PWM files are displayed, click on the one you wish to select.
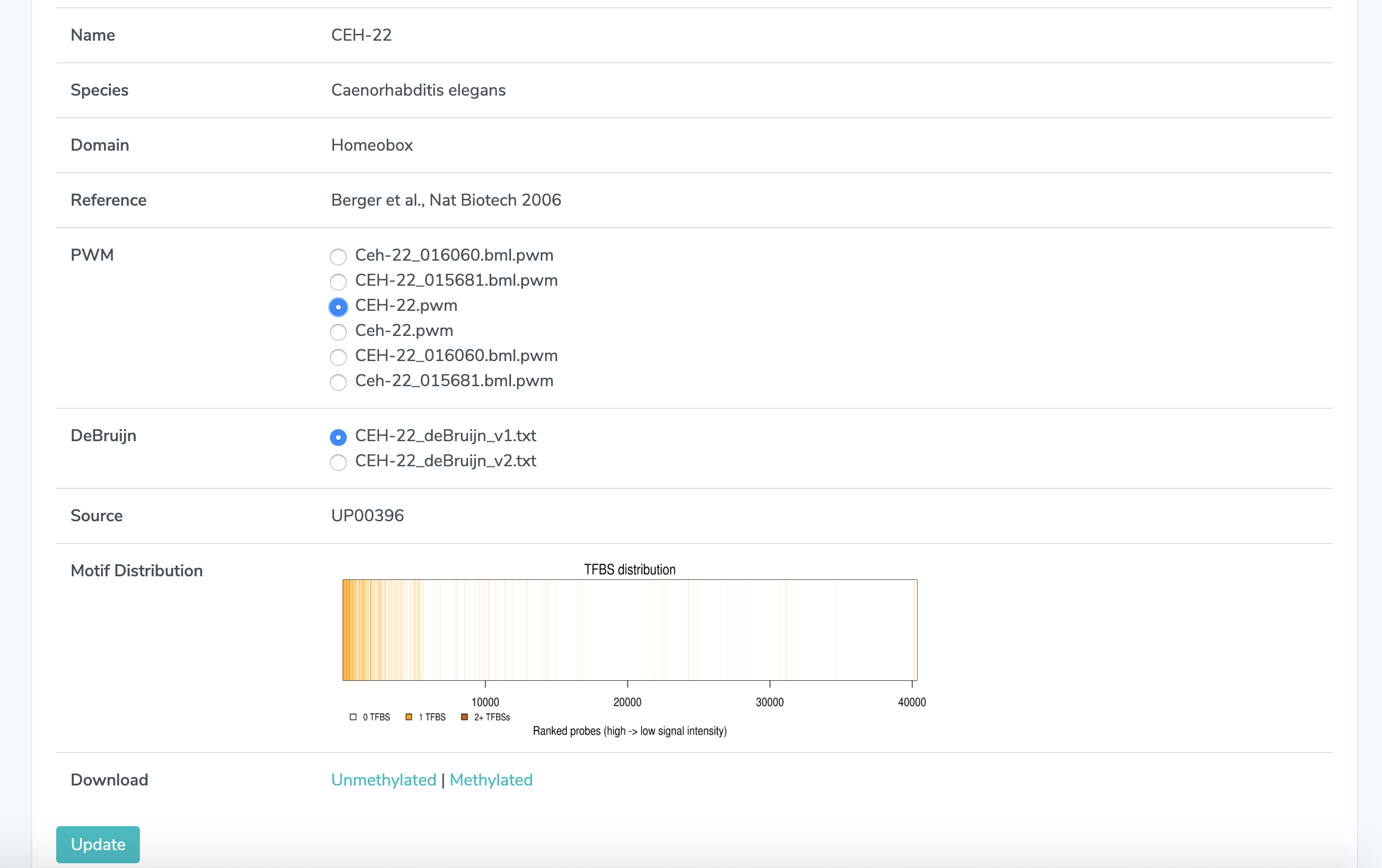
2. DeBruijn Selection If multiple DeBruijn files are displayed, click on the one you wish to select.
3. Click "Update" The heatmaps and boxplots will be updated to reflect your selection.
Comparing Two Transcription Factors
1.1 Select a Database Click on "Compare TFs" in the navigation bar on the left. Once on the comparison page, choose a database to search in for each transcription factor. You may compare the same trancription factor from two different databases if it is available. The options are JASPAR, UniProbe, EpiSELEX-seq, and MeDReaders. EpiSELEX-seq contains in-vitro methylation data. MeDReaders combines in-vivo ChIP-seq and whole-genome bisulfide sequencing data.
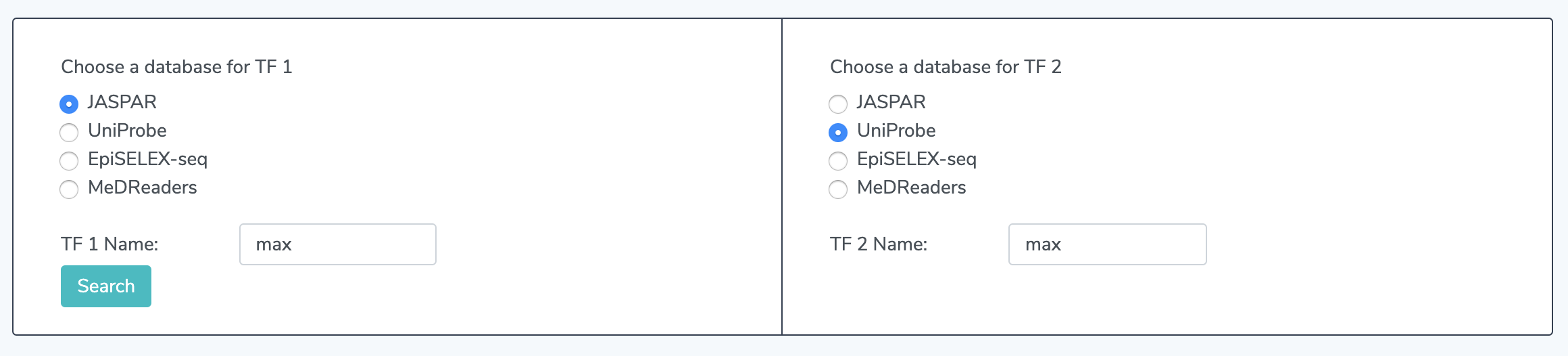
1.2 Enter TF Names Enter names for TF 1 and TF 2. This is required
1.3 Click Search A new form will appear below for you to select your TF's.
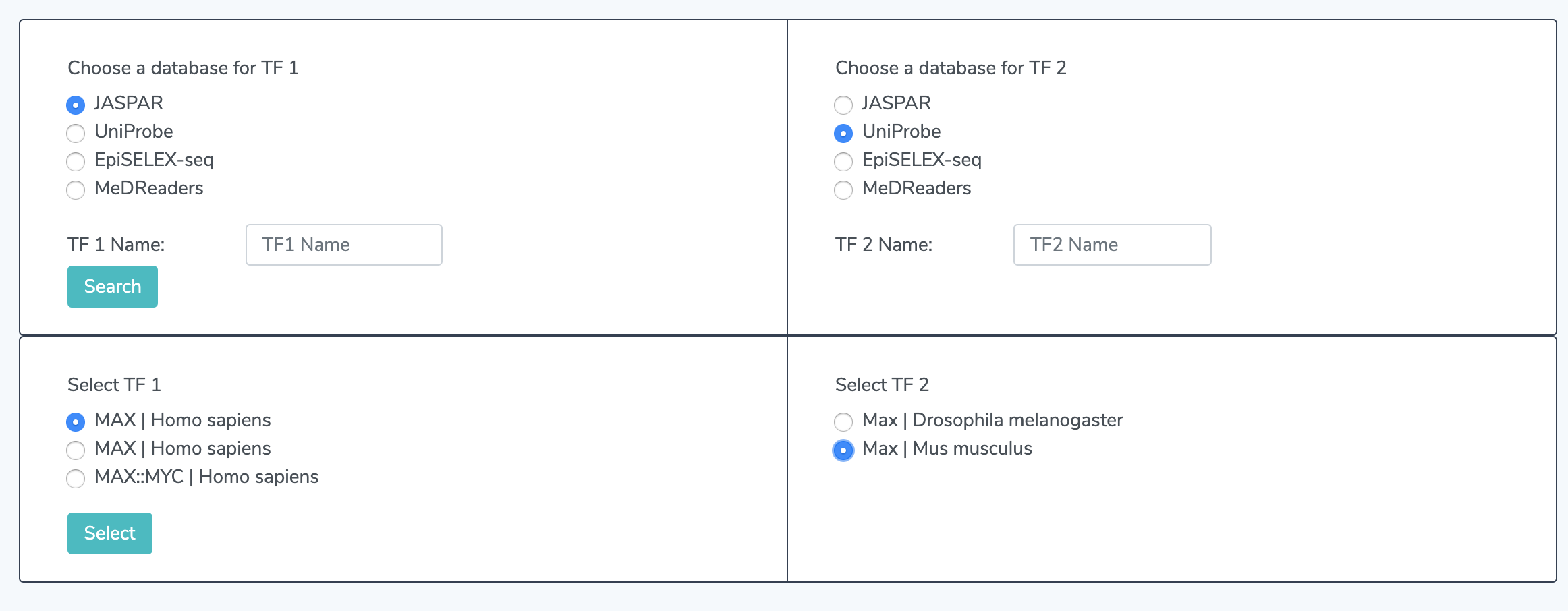
2.1 Select TF Names Select TF 1 and TF 2 from the button options. Multiple TF's may appear if your search returns multiple TF's but only one may appear. If none appear, please verify that the particular TF is available from the selected database using our search page.
2.2 Click Select A new form will appear below for file selection.
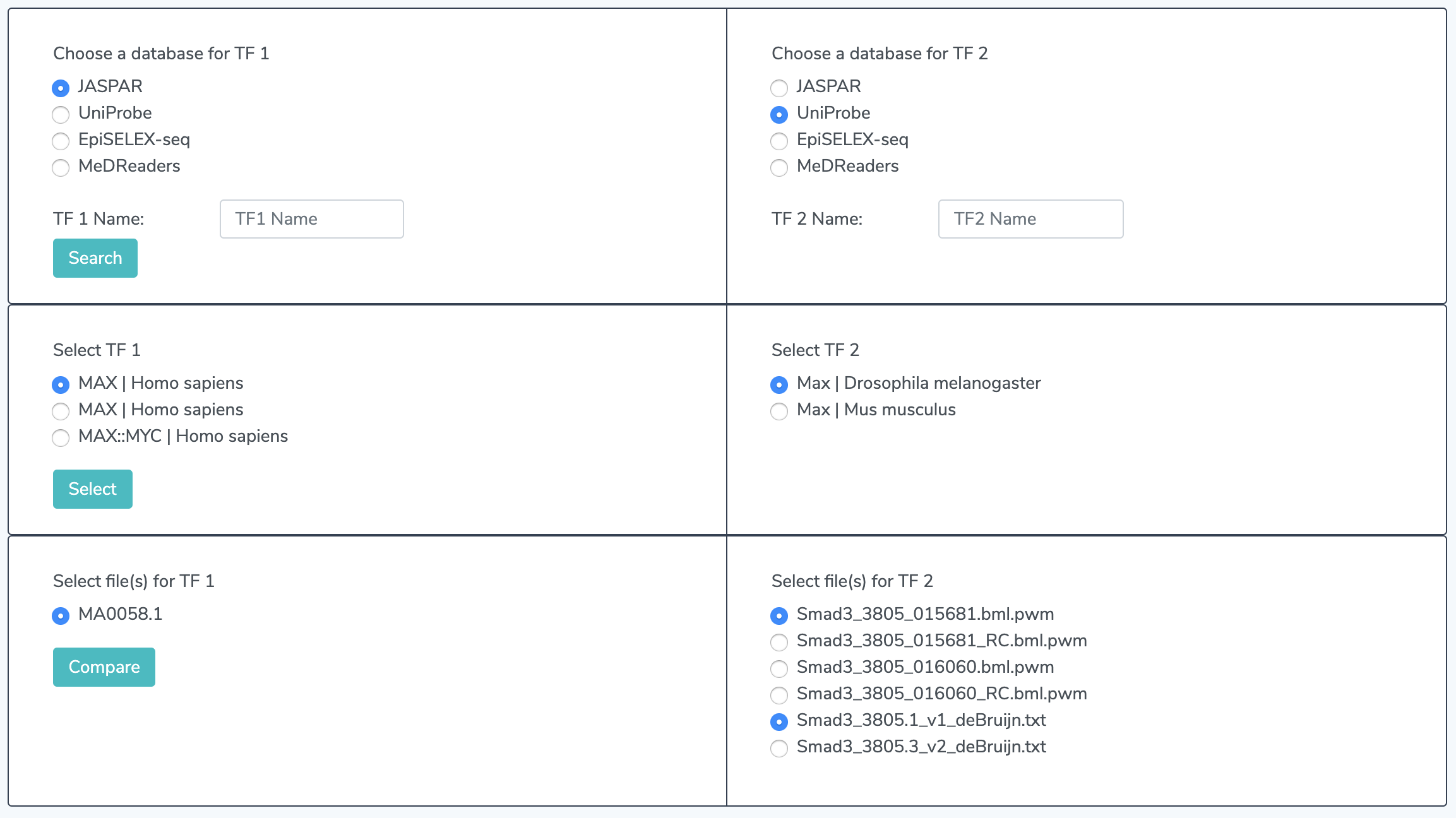
3.1 Select Files Multiple files may appear for one TF. If so, make a selection.
3.2 Click Compare
4.1 Choose Alignment Enter reference positions for alignment or select shape features to auto-align by. Aligning by reference position cannot be done at the same time as alignment by shape. Aligning by shape features will only display comparison heatmaps for the selected features, but aligning by reference will show all shape features.
4.2 Update Alignment Click "Alignment by Reference" or "Align by Shape." Reference position will be updated after alignment by shape.
Using Quick Search
1. The Quick Search Bar At the top of each page is the Quick Search Bar. This search returns results from all four of our sources (JASPAR, UniPROBE, EpiSELEX-seq, and MeDReaders) and can be an easy way to find a TF if you already know the name. It will tell you which of the four databases have entries corresponding to that TF name. Simply type in the name of the trancsription factor you are searching for and hit enter.
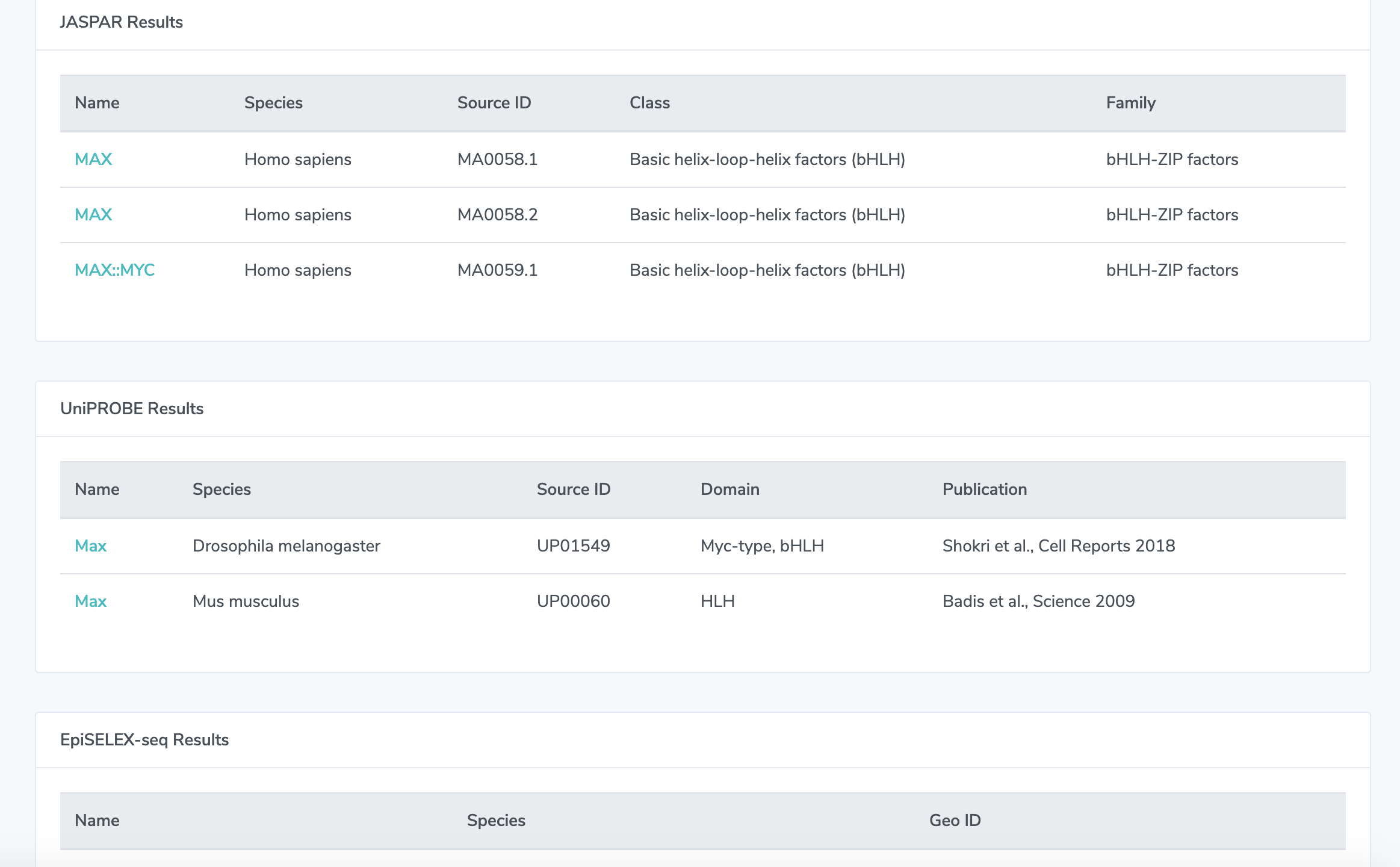
2. Quick Search Results On this page, you will find the results from searching by name in all four databases that we source our data from. If there are no results displayed, then TFBSshape does not have an entry for that transcription factor. Clicking on any of the TF names in the results will take you to the same detail page you would reach through our regular search page.
Mutation Design
1.1 Select a Database and Search a TF Choose which database to search in for your TF. The options are UniPROBE and JASPAR. Then type in the name of your desired TF and click search.
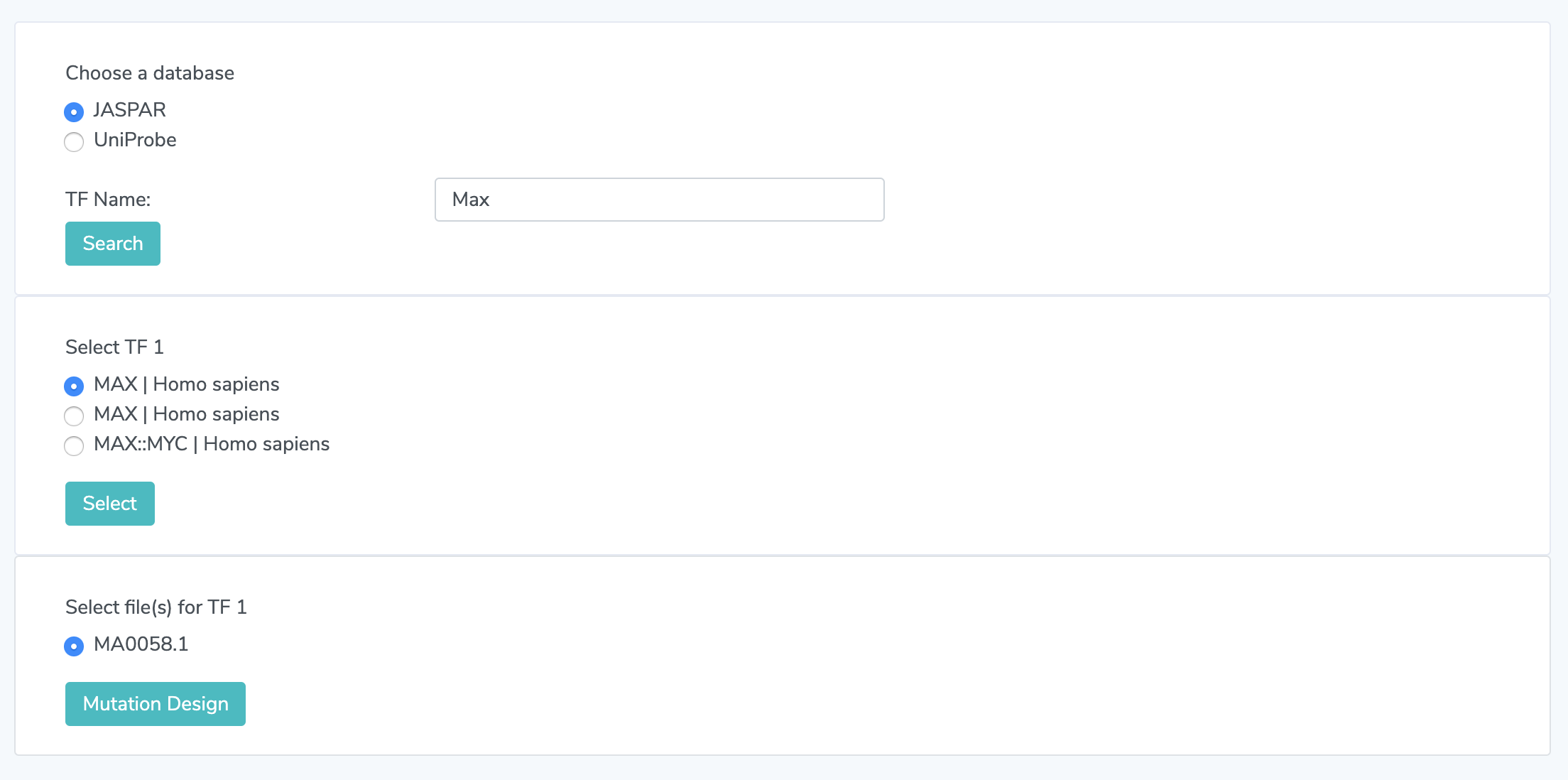
1.2 Select a TF Search results from 1.1 will be displayed. Pick an option. If none are displayed, there are no entries in that database that match the TF name you searched.
1.3 Choose a File. Multiple files may appear for a TF, if so, make a selection.
2. Choose a Sequence Type in the sequence you want to mutate. Lower-case letters indicate those bases can be mutated, upper-case letters indicate they are fixed. The sequence should have the same length as the those binding sites selected in 1.3. In this example, it must be 14 bases in length.
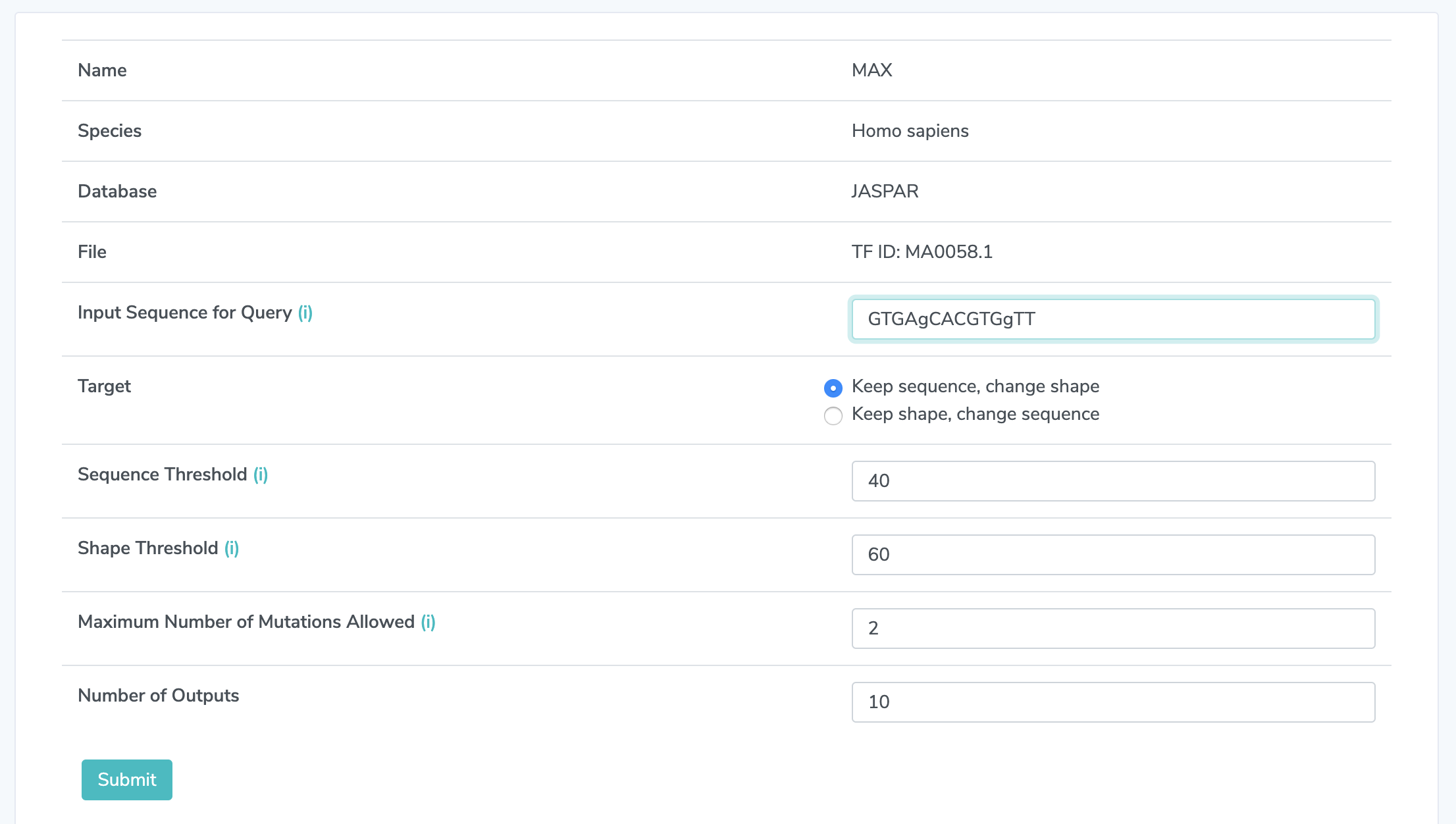
3.1 Choose a Target Choose "keep sequence, change shape" or "keep shape, change sequence." This specifies the goal for the mutation design tool.
3.2 Set Sequence Threshold Choose a number between from 0 to 100. Every mutation has a DNA sequence distance to the input sequence, as well as a percentile rank of the distance relative to the list of distances among all possible mutations. Higher percentile means larger distance. This threshold sets an upper bound of percentile ranks for selected mutations if the target is ‘Keep sequence, change shape’ and a lower bound if the target is ‘Keep shape, change sequence’. The default is 40.
3.3 Set Shape Threshold Choose a number between from 0 to 100. Similarly, every mutation has a DNA shape distance and a percentile rank. This threshold sets a lower bound of percentile ranks for selected mutations if the target is ‘Keep sequence, change shape’ and an upper bound if the target is ‘Keep shape, change sequence. The default is 60.
4. Set the Number of Mutations Allowed This must be an integer greater than 0. If this value exceeds the number of uppercase bases in the input sequence, that number will be treated as the maximum number of mutations allowed. The default is 2.
5. Set the Number of Outputs This sets the maxmimum number of output sequences to be displayed.
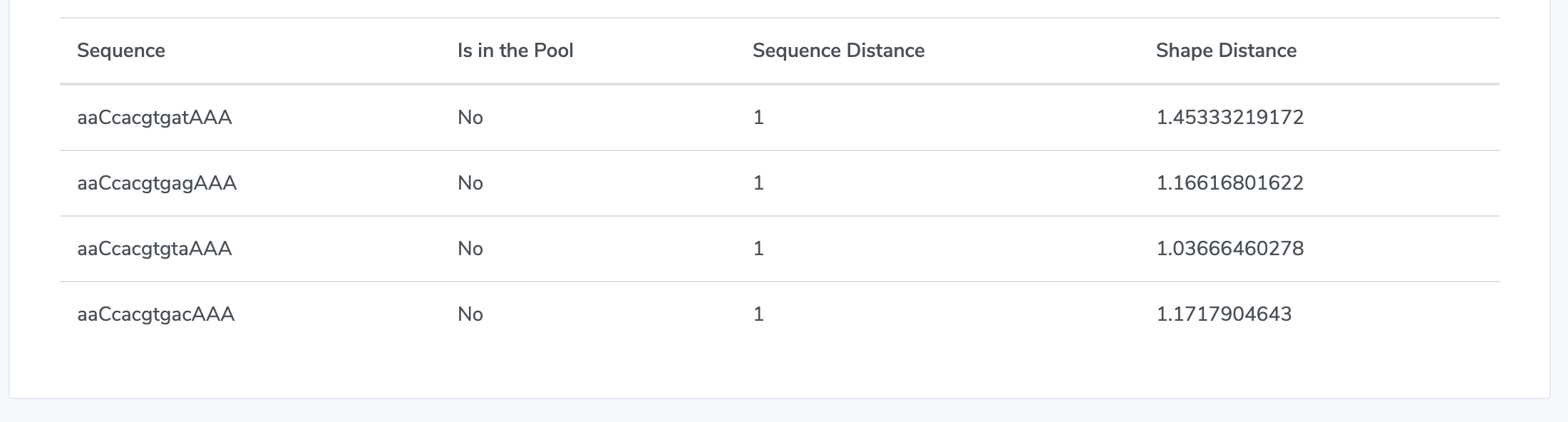
6. Get Results Mutated sequences will be returned in a table. The "Is in the Pool" column indicates whether that mutated sequence is in the pool of binding sites for that TF, according to the database and file you chose in steps 1.1 and 1.3. The distance between wild-type sequence you entered and each mutated sequence is determined by the similarity between the two strings of DNA sequences. This is calculated as Levenshtein distance which counts the number of deletions, insertions, or substitutions required to transform from wild-type to the mutated sequence. Furthermore, to calculate DNA shape distance, shape profiles (MGW, ProT, Roll, and HelT) for the wild type and mutated sequences are first derived with DNAshapeR and are then concatenated as vectors. The distance between these two vectors is represented as Euclidean distance.